
MTS Helpdesk
Question:
What are the elements of the Analytical Procedure Lifecycle, as mentioned in guidance documents such as ICH Q14 and USP <1220>?
Answer:
An analytical procedure (or method) lifecycle consists of the activities associated with the procedure development, validation, routine use, transfer to another laboratory, and change control, throughout the period of time that it is in use.
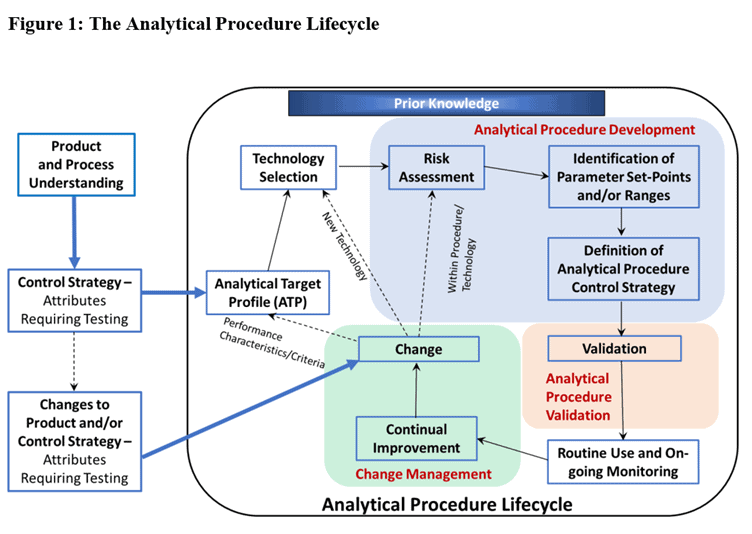
In section 2.2 of the ICH guideline Q14, Analytical Procedure Development, there is a figure which depicts the elements of the analytical procedure lifecycle. This diagram provides an excellent overview of the activities involved and it is described fully in the video below.
Video Transcript:
In section 2.2 of the ICH guideline Q14 on analytical procedure development, there is a figure which depicts elements of the analytical procedure life cycle.
Where do methods start? Well, they start with you identifying that you need one. Where they begin is from your process and your product. You identify that you need to know something, and it could be a final product, or could be the drug substance, and also the process – there might be something you need to know about that. That will define the control strategy for the product and process, and that control strategy will identify that you need this piece of information. That critical quality attribute will usually be that an analytical method is required for it. That’s the starting point of where a method comes from – you identify that you need one.
We’re now in the procedure life cycle and the first thing that is done is this analytical target profile (ATP) and the purpose of the ATP is to say, ‘that’s what we want the method to be able to do’. And that makes sense doesn’t it? Just defining your objective before you begin. Now usually the theoretical approach for this is you decide ‘I need this test’ and then you decide which technology you’re going to use, that would be the more correct way of looking at this. But sometimes you will just directly say ‘oh, I need a HPLC method’ or ‘oh, I need a Karl Fischer method’ you know, that bit sometimes feels a little bit forced. But in theory, you decide what the method needs to do and then you choose an appropriate technology, and that decision is based on your expertise and your prior knowledge. That’s where it’s coming from.
Once you’ve chosen your technology, then we go into the blue box which is the procedure development, and the first thing that we would do is use all of our prior knowledge and perform a bit of a risk assessment to decide what parameters are important here. This is really about your understanding of how the technique works to decide ‘what am I going to change?’, ‘what am I going to move?’. In chromatography it might be the mobile phase, you know, what sort of parameters have I got? You might not always consider that a risk assessment, it might not feel like one, you might not consciously call it that – but that’s basically what you’re doing at that stage. Later on in the process it is more obviously a risk assessment, but for now in the first stage of method development it is what you’re doing.
What you will do then is identify set points for the various parameters in your method, and/or ranges for the parameters in the method. That’s really where you’re doing experiments, there’s different ways of doing them, you might do a one-by-one approach, a univariate approach, or you might do a multivariate approach where you try lots of different parameters at once, and you do screening studies, you run those to get an idea of what’s working, what isn’t working, then you follow it up with another screening study where you’ve changed the parameters based on the output from the first experiment. What you end up with then is a method, you’re going to define that procedure, it’s the last box there.
That method could have set points for the parameters and traditionally that’s usually what we would have. It could be a temperature, maybe that temperature is to do with sample extraction, or it could be a column oven, or it could be a control of your samples to keep them refrigerated. Temperature is a nice parameter that you might have to find in the method. But you might know that actually as long as it’s between this range it’s okay, and that’s what’s coming out of your method development, you might prefer to have a range for it and that’s where these newer ideas about this regulatory flexibility – having ranges in your methods instead of set points comes in.
Definition of the control strategy is a big picture, it’s about what the method is, how it’s used, how it’s written, what the parameters are, but also the system suitability. You want to have at least a prototype system suitability.
I think once you’ve gone through validation it would be quite reasonable to revisit it and make sure it’s adequate, but you should have it before you go into validation.
That’s us, we’ve got our method, we’re ready to validate it, we go down then into the beige box to validation. And that’s Q2. That’s just done, could be quite a lengthy study, but now we’ll just move through it quickly. Now we’re ready to put this method into routine use, so it can go to into a QC environment for example. The next little bit is ongoing monitoring. You might do this, or you might not, it is not a GMP requirement so in a QC environment it could be there, but it might not be. I think it’s only a matter of time before it is a QC requirement, a GMP requirement. A lot of us monitor methods so that we can get feedback loops, and we can see if the method is performing as expected. If you do that then it’s possible you could go into the next box. Because you can see the arrow going from ongoing monitoring is into the change management box, and in particular, continual improvement.
If you don’t trend and monitor your methods, you haven’t really got as easy a way of identifying an opportunity for continual improvement. What would trending look like? There are different options on this. There is some good information in USP <1220>. What sort of things might we look at? System suitability is a great one. You might log all of your system suitability, or you might just keep a trend of the ones that fail. Invalidated system suitabilities can tell you a lot about how the method’s working, If you’re having a lot of them, and if it’s always the same reason, it might indicate an issue with the method. The other one that we always do because we’re required to is trending on OOS results. If your method is in a QC environment, you would do OOS investigations and you have to trend those. That trending should identify if there’s a method issue. These are a couple of examples, and they’re very commonly done I find in biopharm labs, but possibly not as commonly in small molecule labs, but it’s growing. I think where it came from in biolabs is the bioassay. You really need to trend that method. People use statistical control for that, they do control charts on it. It’s difficult, so having that method information and that continual improvement loop is so crucial there, and then people realized, this is really good, and expanded it to other sorts of methods so it’s quite common and it would be considered best practice.
That’s one source of change management. If we go up the box you can see there in that continual improvement, you might recognize that there’s a change you want to make to the method, and the other place a change could come from is outside of the methodology, so we follow this arrow backwards and you can see it’s right back out at what you’re testing, the sample itself, the test article. If something changes to the product, maybe the formulation changes, or something changes to the process, which could affect your profile, it could affect your in-process control testing, those sorts of things could really change your method. They’re all our possibilities for change. If we were in continual improvement the sort of change it might be could be fairly minor. It could be a bit of a tweak within the original method. That’s this first line here. Maybe you want to make a little tweak to your method, you think, well those conditions would be better if I put the set point a little bit differently. What you’d need to do within that is a risk assessment of what does that affect, and is that within what I have approved? Where are my risks? And then you might decide it doesn’t change anything, it’s very minor, it’s absolutely fine, I don’t have to do any experiments, it’s in my design space, I’ve already approved that all, it’s all fine. Or you might go through and say well it could affect something, so you might actually have to define what experiments are required, you’re going through the validation box again. But it could be like a partial validation not a complete revalidation and then you’re back into routine use. That could also happen if you change the product. If it’s quite a minor change, you might realize that’s not going to affect the method. You go through the different parameters and you say, it’s not going to affect my accuracy, my precision, my calibration, and so on. This is where your understanding comes in. It could be a much bigger change. If it’s coming from outside, if it’s coming from changes to the product or the control strategy, you might actually identify you need a new method, or in an existing method the specification needs changed. That could change the ATP. We could be coming in this direction, brand new ATP, a brand new technology selection, brand new method, it goes through the whole process in full, it’s a full method development and a full validation before you go to routine use. Or it could be that your change just requires new technology but not a new ATP, but again it’s a full method validation before it can go into routine use.
Submit a Question to the MTS Helpdesk
Do you have a question for our expert relating to the topic of analytical chemistry? Use our enquiry form to send it to us.
Related content from our Resources Library
Services related to this content that we offer
A full discussion of analytical procedure lifecycle management is included in our courses, ‘Validation, Verification and Transfer of Methods for Pharmaceutical Analysis‘, and ‘Validation, Verification and Transfer of Methods for Biopharmaceutical Analysis‘.
Visit our courses page for a full list of available courses and the schedule of currently available course dates.